Welcome to LigQ
The discovery of small molecules with biological activity, is of great interest for the development of therapeutic agents. One problem is how to narrow the search of possibly active compounds from large ligand databases.
Starting from a selected protein target, our aim is to select a small set of potential drug-like ligands adequate for performing a Virtual Screening(VS) procedure.
In this application you would perform the following operations clicking in "New Calculation":
Over the option "New Full Pipeline" all this calculations are computed in a pipeline, using the output of the previous as the inputof the following. Also, a full download of the protein structure, the candidate binding grid and all the compound's three dimensional structuresare packed to run a docking in rDock or AutoDock softwares.
Starting from a selected protein target, our aim is to select a small set of potential drug-like ligands adequate for performing a Virtual Screening(VS) procedure.
In this application you would perform the following operations clicking in "New Calculation":
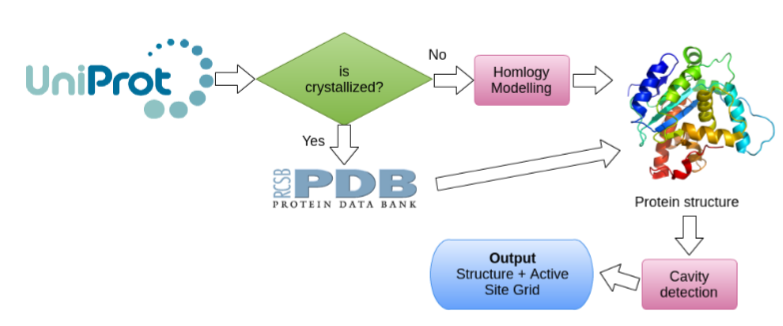
Pocket Detection Module
The aim of this step in the pipeline is to find those regions that arecandidates of been binding regions of drug-like compounds.If an UniProt accession is given, then his best structure is selected.If the protein has not been crystallized then an homology model is built. If a PDB code is given then that structure is selected. Over the final structure a pocket detection software is used to compute the candidate region(s) of binding.
Ligand Detection Module
The aim is to build a set of compounds with evidence of binding to the input protein or similars. This evidence may come from crystallization experiments and/or from biological assays with measured affinity. ChEMBL and PubChem databases have a flag in their data structure to point out that some compound is active or not in a BioAssay over certain protein, local versions of this databases were mounted to perform the queries. The results could be extended with a search over the proteins in the same PFAM family of the target protein.

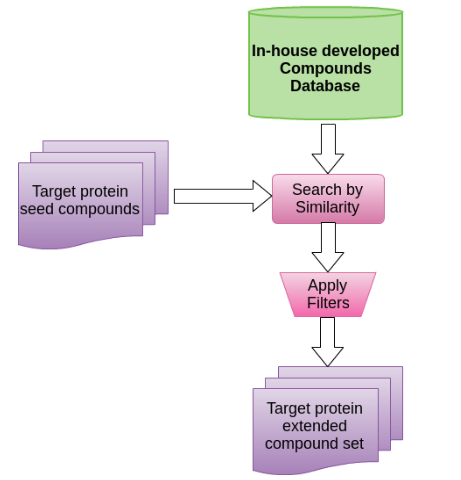
Extend Ligand Set Module
Having one or more input compounds (the server accepts ZINC code, PDB compound code, InChi or SMILES) a query is performed over large compound databases to look forward to similar compounds based in 2D fingerprints and phisico-chemical properties similarity.
Ligand Structure Generation Module
Given one or more compounds in any two dimensional format, all his valid three dimensional structures are computed. To perform this calculations stereoisomers, isomers and microspecies with high probability of appearance are computed. After that, for each specie his geometry are computed.
